

A groundbreaking study published in Bone Research on May 29, 2025, unveils novel insights into the molecular orchestration of skull development, highlighting the indispensable role of RUNX2 in cranial base growth. The cranial base, forming the foundational floor supporting the brain and upper airway, develops through a process critically dependent on synchondroses—specialized cartilage growth plates consisting of organized layers of chondrocytes. These cells undergo a tightly regulated transition from cartilage to bone, enabling the skull to elongate appropriately from front to back. Disruptions in this process have been linked to several craniofacial malformations, such as skeletal Class III malocclusion and foramen magnum stenosis, both of which severely impact fundamental functions like breathing, mastication, and speech.
Despite longstanding recognition of RUNX2 as a pivotal transcription factor governing osteogenesis, the detailed mechanistic role that RUNX2 plays within cranial base synchondrosis chondrocytes has remained enigmatic. Prior research associated RUNX2 deficiency with cleidocranial dysplasia—a congenital disorder characterized by delayed bone development affecting facial structures and skull base—but a comprehensive understanding of its specific cellular and molecular contributions to cranial synchondroses was lacking. This gap has impeded deeper insight into how RUNX2 interacts with key signaling pathways regulating cranial bone development.
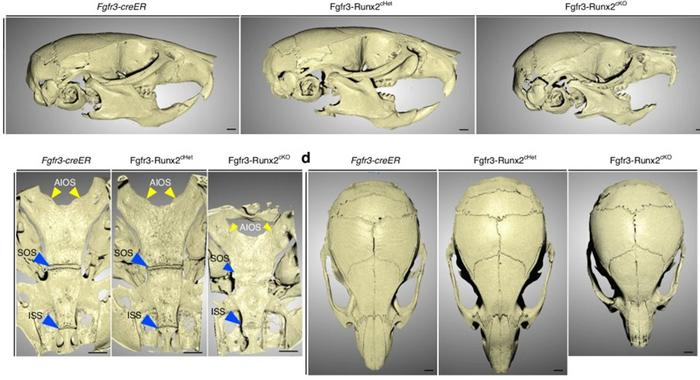
Addressing this critical knowledge deficit, researchers from the University of Texas Health Science Center at Houston School of Dentistry and the University of Michigan School of Dentistry employed a sophisticated tamoxifen-inducible genetic mouse model that selectively ablates RUNX2 expression postnatally in a targeted population of synchondrosis chondrocytes expressing the Fibroblast Growth Factor Receptor 3 (FGFR3). This inducible system, combined with fluorescent lineage tracing, allowed the team to meticulously monitor the fate, morphology, and proliferation dynamics of RUNX2-deficient chondrocytes during crucial postnatal cranial base growth phases.
.adsslot_tyYzvDO7Cb{ width:728px !important; height:90px !important; }
@media (max-width:1199px) { .adsslot_tyYzvDO7Cb{ width:468px !important; height:60px !important; } }
@media (max-width:767px) { .adsslot_tyYzvDO7Cb{ width:320px !important; height:50px !important; } }
ADVERTISEMENT
High-resolution micro-computed tomography (micro-CT) scanning of these mice revealed stark craniofacial phenotypes in the absence of RUNX2, including pronounced skeletal dwarfism and significant shortening of the cranial base. Histological analyses showed premature ossification within the synchondroses, which correlated with disrupted chondrocyte organization. Specifically, RUNX2 loss led to disarrayed chondrocyte layers, diminished proliferative capacity, and increased apoptotic cell death. These findings underscore that RUNX2 is vital for maintaining the structural and functional integrity of the cartilage growth plates that drive normal skull elongation.
Of particular mechanistic interest, lineage tracing experiments elucidated that chondrocytes lacking RUNX2 failed to adequately differentiate into osteoblasts, the bone-forming cells essential for ossification. This impaired differentiation trajectory offers a cellular explanation for the premature closure and ossification defects observed at the tissue level. It suggests that RUNX2’s function extends beyond its established role in bone cells to critically govern the balance between cartilage maintenance and transition to bone within the cranial base.
Furthermore, the study discovered an unexpected molecular crosstalk between RUNX2 and the FGFR3 signaling pathway, a known regulator of cranial development. RUNX2-deficient chondrocytes exhibited markedly elevated expression levels of FGFR3 and downstream MAPK signaling components. This overactivation implies that RUNX2 normally serves to suppress FGFR3-mediated signaling within synchondrosis chondrocytes, helping finely tune cartilage proliferation and differentiation. Dysregulation of this RUNX2-FGFR3 axis appears to disrupt the delicate equilibrium required for sustained cranial base growth.
Professor Noriaki Ono, who led the investigation, emphasized the significance of these findings: “Our research reveals a novel regulatory axis wherein RUNX2 modulates FGFR3 activity in vivo, a dynamic previously inferred only from in vitro models. This insight provides a foundational understanding of the molecular interplay directing skull base formation and imparts new perspectives on the pathogenesis of craniofacial malformations.”
The implications of this research extend beyond basic developmental biology. By delineating the molecular mechanisms underpinning cranial base growth and elucidating RUNX2’s multifaceted role, the study opens exciting avenues for therapeutic interventions aimed at craniofacial dysplasias. Targeting the RUNX2-FGFR3 signaling pathway promises novel approaches to ameliorate bone growth defects, potentially benefiting patients with genetic disorders affecting skull development.
Importantly, the use of an elegant genetic approach combining inducible gene deletion and in vivo lineage analysis exemplifies the power of contemporary mouse models to unravel complex bone biology. This methodology permits temporal and spatial precision in dissecting gene function within intricate cellular microenvironments, offering a blueprint for future studies investigating skeletal development and disease.
Taken collectively, this work not only reinforces RUNX2’s stature as a master regulator of bone formation but also redefines its role within the unique context of cranial synchondroses. It affirms that synchronous regulation of cartilage proliferation, differentiation, and ossification is essential for proper cranial architecture, and that disruption of this balance leads to profound anatomical and functional impairments.
Future research scrutinizing downstream effectors of the RUNX2-FGFR3 axis and their interplay with additional signaling pathways will be critical to fully decode the molecular networks governing skull morphogenesis. Such insights will accelerate the development of precision medicine strategies tailored to correct or prevent craniofacial defects from infancy through adulthood.
In summary, this study delivers a transformative leap in understanding the cellular and molecular bases of cranial base development, situating RUNX2 as a key sentinel maintaining cartilage integrity and coordinating growth factor signaling. These findings bear immediate relevance for clinical and translational research aimed at tackling cranial skeletal malformations, underscoring the promise of targeted modulation of bone regulatory pathways in craniofacial therapeutics.
Subject of Research: Animals
Article Title: RUNX2 is essential for maintaining synchondrosis chondrocytes and cranial base growth
News Publication Date: 29-May-2025
Web References: http://dx.doi.org/10.1038/s41413-025-00426-z
References: DOI: 10.1038/s41413-025-00426-z
Image Credits: Dr. Shawn A. Hallett and Renny T. Franceschi from University of Michigan School of Dentistry
Keywords: Bones, Life sciences, Organismal biology, Anatomy, Human anatomy, Musculoskeletal system, Skeleton, Skull, Cranium, Health and medicine, Health care, Bone diseases
Tags: Bone Research study findingschondrocytes transition to bonecleidocranial dysplasia insightscranial base growth mechanismscraniofacial malformations causesforamen magnum stenosis consequencesmolecular orchestration of cranial developmentRUNX2 role in skull developmentsignaling pathways in bone developmentskeletal Class III malocclusion impactssynchondroses cartilage growthtranscription factors in osteogenesis

