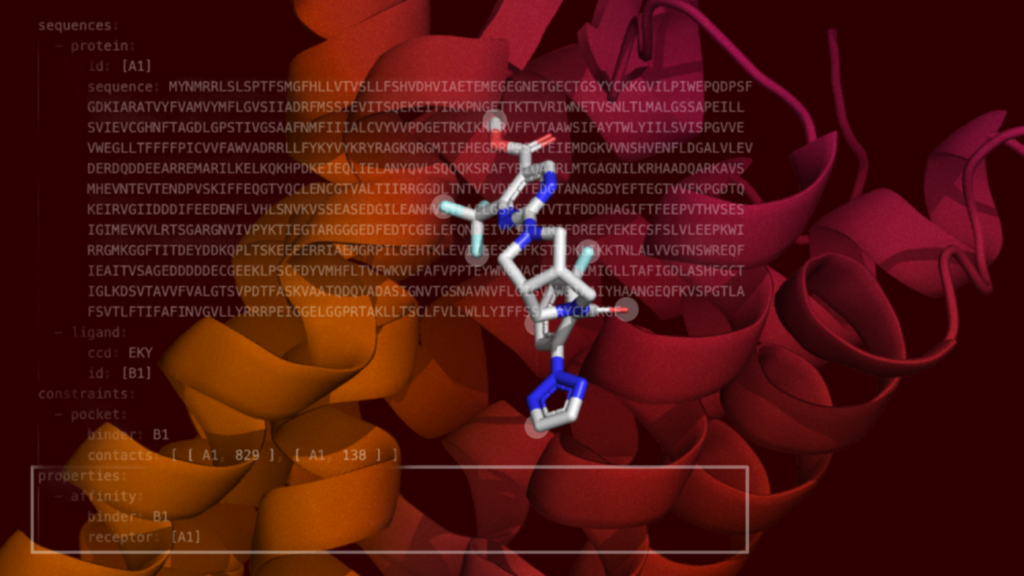
![A small molecule binds to an OX2 protein. [Recursion]](https://www.genengnews.com/wp-content/uploads/2025/06/OX2-with-code-2-1024x576.png)
Researchers from the Massachusetts Institute of Technology (MIT) Jameel Clinic for Machine Learning in Health have announced the open-source release of Boltz-2, which now predicts molecular binding affinity at newfound speed and accuracy to democratize commercial drug discovery. The model is available under the highly permissive MIT license, which allows commercial drug developers to use the model internally and apply their own proprietary data.
The work was done in collaboration with Recursion, the Salt Lake City-based artificial intelligence (AI) drug discovery company that combined with Exscientia last year, and is posted as a preprint on the MIT researchers’ website. (It has not yet been peer reviewed.) Lead authors of the preprint include Saro Passaro, Gabriele Corso, PhD, and Jeremy Wohlwend, PhD, from the lab of Regina Barzilay, PhD, distinguished professor of AI & health at MIT.
The model’s predecessor, Boltz-1, was released last November as the first fully open-source model to achieve AlphaFold 3-level accuracy in predicting the 3D structure of biomolecular complexes. Boltz-1 quickly became one of the most widely adopted co-folding models in the industry, with more than 200 biotech companies applying the model in their pipelines, according to MIT. Boltz-1 has also gathered a growing collaborative Slack community of more than 1,300 developers who build upon the model, share improvements, and discuss new applications.
Lifts all boats
In May 2024, Demis Hassabis, PhD, co-founder and CEO of Google DeepMind and Isomorphic Labs, took to social media to declare a groundbreaking next step for drug discovery—the ability to “predict the structures and interactions of nearly all of life’s molecules with state-of-the-art accuracy.”
Hassabis and colleagues had just published AlphaFold 3 in Nature. The latest update to the soon-to-be Nobel Prize in Chemistry recognized protein structure prediction algorithm expanded the model’s capabilities to a broad spectrum of biomolecular interactions, including small molecules, DNA, RNA, and more.
For the field of drug discovery, which faces a mediocre 10% success rate, hitting the right target with atomic precision to achieve therapeutic effect remains the core challenge. While today’s R&D pipelines are dependent on resource-intensive experimental screens to identify promising drug leads, achieving accurate in silico prediction of molecular interactions, as promised by AlphaFold 3, would identify top candidates much earlier in the game, significantly cutting timelines and saving costs.
Yet DeepMind’s claims to revolutionize drug discovery were not mutually shared. In contrast to the launch of AlphaFold 2 in 2021, Nature’s publication of AlphaFold 3 lacked the open-source code, preventing academic and industry researchers from applying the model to their own drug discovery efforts. The omission led to a protest letter signed by more than 1,000 scientists calling for AlphaFold 3’s transparency.
While AlphaFold 3 developers countered the outcry with promises to release the code within six months (a pledge that was later kept but under a non-commercial license), that did not stop MIT researchers from cooking up a model of their own.
Najat Khan, PhD, chief R&D officer and chief commercial officer at Recursion, stated the open-source release of Boltz-2 “lifts all boats” in making progress in the integration of tech, biology, and chemistry.
“Binding affinity is core to developing a therapeutic start to finish and has been the fundamental issue that a lot of us have been trying to grapple [with],” Khan said in a press briefing. “The value of this collaboration is significant technological advancement geared to the purpose of application, which is drug discovery.”
Barzilay said that binding affinity has been an open problem in drug discovery for decades, and that only novel machine learning techniques were able to solve. “This is not only an advancement in life science research, but also an important discovery and advancement in machine learning and computer science,” she said.
Barzilay also highlighted that another important feature of Boltz-2 is achieving a better understanding of core biological phenomena and mechanisms of action, which is crucial for both discovery and regulatory approval for a drug.
Corso emphasizes the importance of model accessibility as “99% of drug developers are outside of companies like Isomorphic Labs.” He said the biggest reward from releasing Boltz-1 was seeing the community rally behind an open-source project.
“Just at a time where it seemed inevitable that closed models like AlphaFold 3 would dominate the field, many researchers from academia and industry decided to contribute to an open-source project like Boltz to build new capabilities and open them up for everyone to use,” Corso told GEN Edge.
Let it bind
Binding affinity measures the strength of interaction between a drug and its target and is a key drug discovery metric that can dictate the progression of a candidate through the development pipeline from “hit discovery” to “lead optimization.” While AlphaFold 3 made the advance of accurately predicting molecular complex structures, in silico binding affinity calculations as achieved by Boltz-2, had not been (publicly) shown by DeepMind and Isomorphic Labs.
Both AlphaFold 3 and Boltz-1 were trained on the protein data bank (PDB), the public repository housing more than 200,000 entries for experimentally determined protein and nucleic acid structure data. To adapt Boltz-2 to binding affinity predictions, the training set was expanded into new territories, including molecular dynamics simulations, such as MISATO, mdCATH, and ATLAS, and experimental binding affinity databases, such as PubChem and ChEMBL.
In terms of accuracy, Boltz-2 was the leading affinity performer at the December 2024 Critical Assessment of protein Structure Prediction 16 (CASP16) competition, the biannual experiment that assesses the latest state-of-the-art models in structural biology. In speed, Boltz-2 is reported to calculate binding affinity values in just 20 seconds, 1000x times faster than the current physics-based computational standard, free-energy perturbation (FEP) simulations.
Given that binding affinity experiments can cost hundreds or thousands of dollars per individual molecule, cost savings can come from applying Boltz-2 to cut down on the time and number of experimental rounds needed to advance a drug candidate.
According to Khan, Recursion already uses Boltz-1 in combination with its own generative AI models. She emphasized that Recursion has seen examples of late discovery programs that were able to be completed within 18 months instead of the industry average of 42 months.
As part of the collaboration, Recursion tested Boltz-2 binding affinity against their internal drug discovery data and developed additional machine learning and physics-based benchmarks to ensure its accuracy. The company looks forward to more discovery examples to come with Boltz-2.
Gain control
After the release of Boltz-1, Wohlwend said feedback from the community expressed a desire for more control of model predictions, including the ability for companies to leverage their internal data without having to retrain the model.
To that end, Boltz-2 introduces templating, where proprietary molecular structures can improve the capacity of the model to predict a particular target, and contact and pocket conditioning, where a known binding location on a protein can assist the model in landing a ligand into a structural pocket.
While Boltz-2 is currently positioned to tackle small molecule drug discovery, Barzilay added that one of the biggest advantages of open sourcing Boltz-2 is the release of the training code, which allows drug developers to fine-tune the model to specific modalities of interest to expand generalizability, a capability that was not possible with AlphaFold 3 given its limited accessibility. According to Barzilay, many pharmaceutical collaborators are already investing resources to apply the model to do this exact task.
When asked whether Boltz-2 was capable of eliminating physical experiments, Khan stated that experimental validation remains important.
“If you capture the data, the wet and dry lab-in-a-loop can actually improve the algorithms as you learn from the experimental validation,” said Khan. She calls for a need for high-quality fit-for-purpose data sets that continue to improve models and performance.
Once Boltz-2 goes live, Corso told GEN Edge he expects to once again see contributions from across the industry to further build upon the model. Corso also looks forward to how practitioners will use Boltz-2’s scalability to rethink how to design molecular workflows with new creative strategies.
“The road we have ahead will continue to push the boundaries of what is possible to predict in silico even beyond small molecule binding affinity,” said Corso.
Wohlwend said the team has always seen Boltz-1 as a stepping stone rather than the finish line.
“It’s exciting to see Boltz-2 bring us closer to our goal of advancing molecular modeling,” he told GEN Edge. “I hope the model sparks new ideas and gives more biologists the confidence and curiosity to explore the use of machine learning in their own work.”
As clashes between commercial interests and AI collaboration rage on, the field will watch as a new revolution in drug discovery continues to rise from the noise.