
In the relentless pursuit of breakthroughs in pediatric oncology, a promising frontier has emerged involving the intersection of molecular targeted therapies and immunotherapy. Neuroblastoma (NB), a formidable childhood cancer often driven by mutations in the anaplastic lymphoma kinase (ALK) gene, presents profound challenges due to its intrinsic resistance to conventional chemotherapy and its ability to manipulate the tumor microenvironment (TME) to evade immune destruction. Recent research spearheaded by Sugitatsu and colleagues introduces a transformative therapeutic avenue that exploits the synergy between an ALK inhibitor, alectinib, and chimeric antigen receptor (CAR) T-cell therapy targeting disialoganglioside 2 (GD2), a surface molecule ubiquitous in neuroblastoma cells.
Neuroblastoma remains one of the deadliest solid tumors in children, and its heterogeneity complicates treatment paradigms. The ALK mutation subgroup comprises a cohort characterized by aggressive disease and poor prognosis, primarily due to the dual adversaries of intrinsic drug resistance and an immunosuppressive milieu engineered by tumor cells. The TME in ALK-mutant neuroblastoma is often laden with suppressive signals, notably the overexpression of immune checkpoint molecules like programmed death-ligand 1 (PD-L1), which paralyze infiltrating cytotoxic T cells and dampen anti-tumor immune responses. Disrupting this immunosuppressive shield is thus central to improving outcomes in this recalcitrant cancer.
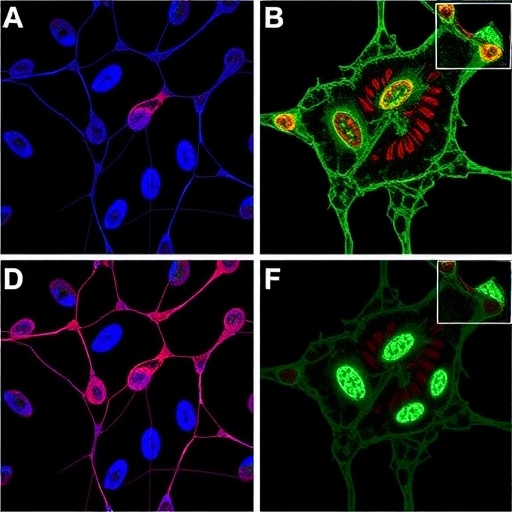
Sugitatsu and collaborators meticulously delineate how combining alectinib, an FDA-approved ALK inhibitor known for its efficacy in ALK-positive non-small cell lung cancer, with GD2-directed CAR T-cell therapy potentiates anti-tumor responses against ALK-mutant neuroblastoma. The novelty lies not only in targeting the oncogenic driver but also in simultaneously dismantling the tumor’s immune-evasive strategies. Mechanistically, alectinib has been shown to downregulate PD-L1 expression on neuroblastoma cells, thereby reinvigorating CAR T-cell activity by relieving inhibitory checkpoint constraints.
The elegant study hinges on unraveling the crosstalk between oncogenic signaling pathways and immune checkpoints. ALK tyrosine kinase activity intricately modulates intracellular signaling cascades that upregulate PD-L1 transcription and surface expression. By inhibiting ALK, alectinib disrupts this axis, effectively reducing PD-L1 levels and lowering the threshold for T-cell mediated cytotoxicity. This molecular disruption enhances the functional persistence and cytolytic potential of GD2 CAR T cells, which are specifically engineered to recognize and lyse neuroblastoma cells expressing disialoganglioside 2.
Understanding the implications of the TME is crucial in appreciating the significance of these findings. Neuroblastoma’s microenvironment has been notoriously immunosuppressive, featuring a constellation of inhibitory cytokines, regulatory T cells, myeloid-derived suppressor cells, and checkpoint ligands like PD-L1. Such complexity not only hampers endogenous immune surveillance but also curtails the efficacy of adoptive cell therapies. By harnessing alectinib to reduce PD-L1 expression, the researchers essentially reconfigure the tumor’s immune landscape to one that is more permissive to CAR T-cell infiltration and memory formation.
Beyond the phenotypic modulation of neuroblastoma cells, the study also explores the functional ramifications for GD2 CAR T cells. Preclinical models demonstrate that these engineered lymphocytes exhibit superior proliferative capacity, enhanced cytokine secretion, and increased tumor cell killing in the presence of alectinib. The combinatorial approach translates into sustained tumor regression and, importantly, delays or prevents relapse, a common pitfall in aggressive neuroblastoma treatments.
In addition, the research addresses a critical bottleneck of CAR T-cell therapy in solid tumors: the immune checkpoint-induced exhaustion of effector T cells. The PD-1/PD-L1 axis has been cited as a primary driver of T-cell exhaustion and apoptosis within tumors. While immune checkpoint inhibitors targeting PD-1/PD-L1 have revolutionized therapies in adult solid tumors, their application in pediatric malignancies has been limited by variable responses and toxicity concerns. By using alectinib as a molecular modulator to suppress PD-L1, the need for systemic immune checkpoint blockade might be reduced, potentially minimizing immune-related adverse effects and offering a tailored treatment modality.
The implications of these discoveries extend beyond neuroblastoma. They exemplify a broader paradigm shift where precision oncology converges with immunotherapy to tackle cancers previously deemed refractory. The integration of kinase inhibitors to sensitize tumors to immune elimination could pave the way for novel combinatorial regimens across diverse malignancies characterized by driver mutations and immune suppression.
Moreover, this dual-targeted strategy highlights the importance of biomarker-driven therapeutic design. The identification of ALK mutations as a predictive biomarker offers clinicians a rational basis to stratify patients most likely to benefit from this combined approach. The use of GD2 as a tumor antigen further fine-tunes the specificity of CAR T cells, reducing off-tumor toxicity and enhancing safety profiles.
While the preclinical data is compelling, translating these results into clinical practice will necessitate carefully designed trials to assess dosing, timing, and potential synergistic toxicities. Monitoring PD-L1 modulation in patient tumors during therapy could provide valuable correlative insights to optimize treatment efficacy. Emphasizing multidisciplinary collaboration among oncologists, immunologists, and molecular biologists will be critical in advancing this therapeutic frontier.
Furthermore, addressing the phenotypic plasticity of neuroblastoma cells and potential resistance mechanisms remains a priority. Tumors may adapt to ALK inhibition or CAR T-cell pressure by altering antigen expression or deploying alternative immune evasion pathways. Continuous innovation in CAR design, possibly integrating multi-antigen targeting or armored CARs resistant to exhaustion, could enhance durability of responses.
Remarkably, this research injects renewed optimism into the quest for curative therapies for high-risk neuroblastoma patients, who currently experience significant morbidity and mortality despite intensive multimodal treatments. The possibility of combining targeted small molecule inhibitors with sophisticated cellular therapies embodies the cutting edge of personalized medicine.
In summation, the study by Sugitatsu et al. charts a compelling course in neuroblastoma treatment by demonstrating how alectinib-mediated suppression of PD-L1 synergizes with GD2 CAR T-cell therapy to overcome tumor immune resistance in ALK-mutated cases. This innovative approach redefines the therapeutic landscape, transforming a historically intractable cancer into one amenable to precise, dual-pronged intervention. As the scientific community continues to unravel the interplay between oncogenic drivers and immune checkpoints, such paradigm-shifting strategies will undoubtedly herald a new era in pediatric oncology, offering hope for durable remissions and improved survival for children afflicted with this devastating disease.
Subject of Research: The study focuses on high-risk ALK-mutant neuroblastoma and investigates the therapeutic synergy between the ALK inhibitor alectinib and GD2-targeted chimeric antigen receptor (CAR) T-cell therapy, particularly through the suppression of PD-L1-mediated immune evasion.
Article Title: Alectinib boosts anti-tumor efficacy of disialoganglioside 2 chimeric antigen receptor T cells in ALK-mutated neuroblastoma by suppressing programmed death-ligand 1.
Article References:
Sugitatsu, Y., Tomida, A., Suematsu, M. et al. Alectinib boosts anti-tumor efficacy of disialoganglioside 2 chimeric antigen receptor T cells in ALK-mutated neuroblastoma by suppressing programmed death-ligand 1. Br J Cancer (2026). https://doi.org/10.1038/s41416-026-03363-8
Image Credits: AI Generated
DOI: 10.1038/s41416-026-03363-8 (Published 23 March 2026)
Tags: Alectinib and CAR T-cell synergy in ALK neuroblastomaGD2-targeted CAR T-cell therapyimmune checkpoint inhibition in pediatric cancerimmunotherapy enhancement with ALK inhibitorsmolecular targeted therapies in childhood cancernovel treatmentsovercoming chemotherapy resistance in ALK-mutant tumorsPD-L1 role in neuroblastoma immune evasionstrategies to overcome immunosuppression in neuroblastomatargeted therapy for pediatric neuroblastomatumor microenvironment modulation in neuroblastoma
