The mutation in the huntingtin (HTT) gene, which causes the incurable, dominantly inherited Huntington’s disease, was identified decades ago, following one of the most closely watched disease gene quests in the early years of gene mapping. But exactly how this triplet repeat expansion mutation functions to cause the disease has eluded researchers—until now.
A team of researchers from the Broad Institute, Harvard Medical School, and McLean Hospital have identified at last how the mutation causes symptom development later in life, opening doors to therapeutic approaches for delaying or preventing development of the disease.
The study reveals that the mutation in the HTT gene is initially innocuous but gradually morphs into a toxic form that causes the death of specific brain cells.
Published in Cell, the study titled, “Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease,” was co-led by Steve McCarroll, PhD, of the Broad Institute, Harvard Medical School, and the Howard Hughes Medical Institute, and Sabina Berretta, MD, of Harvard Medical School and McLean Hospital.
“The point of our work—what we all do—is relieving suffering caused by disease,” said Berretta.
How the mutation works
The HTT gene contains repeats of a codon, CAG, which encodes the amino acid glutamine. Typically, healthy individuals have about 15–35 CAG repeats within the gene; however, in patients with the disease, the repeat sequence expands to more than 40 CAG repeats.
The Boston researchers aimed to better understand how the relative difference in repeats impacts cell health and thus symptom development. “This is a really different way of thinking about how a mutation brings about a disease,” commented McCarroll.
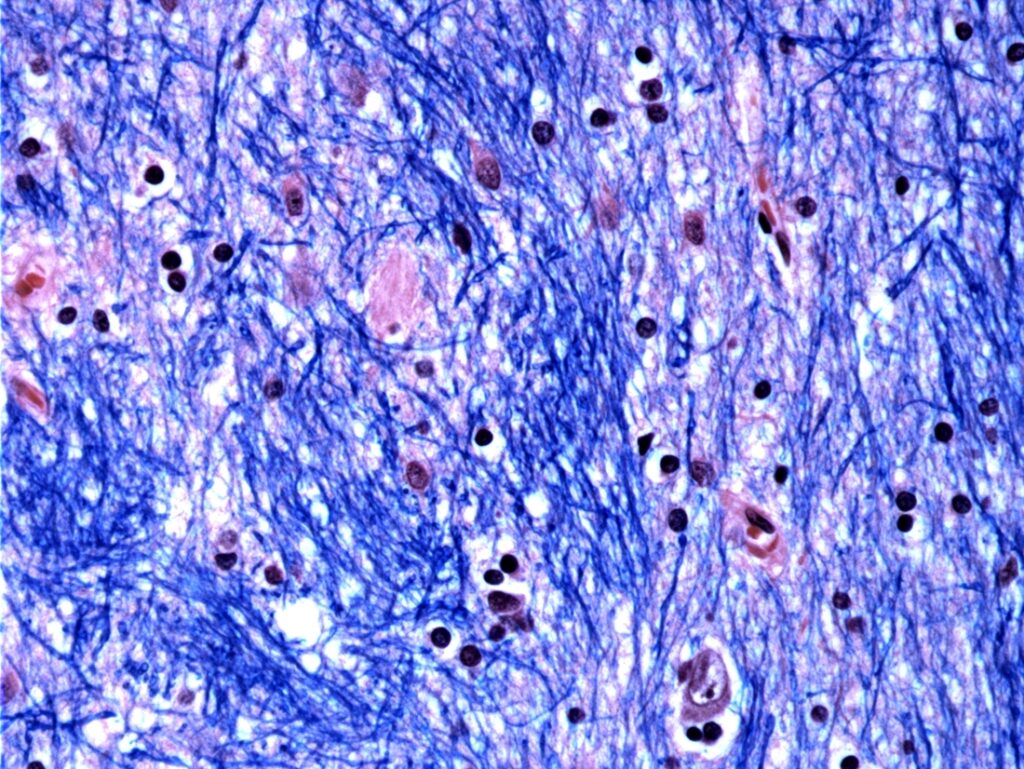
As they describe in their paper, the researchers “developed a single-cell method for measuring this repeat’s length alongside genome-wide RNA expression.” They found that mutated HTT undergoes a process called “somatic expansion,” by which the number of CAG repeats grows, particularly in neurons in the striatum of the brain. Once this number surpasses 150 repeats, the cells become dysfunctional and die.
Using single-cell RNA sequencing, the team studied more than 500,000 cells from brain tissue donated by 53 individuals with Huntington’s disease and 50 controls. Their analysis confirmed that only striatal projection neurons—the cells primarily affected in the disease—develop significant CAG repeats. Furthermore, once the gene in a given cell reaches 80 repeats, which typically takes decades, the accumulation of repeats accelerates, shortening the time it takes to reach 150 repeats, which is on the scale of years.
Due to this inexorable increase in repeats in the mutated gene, the authors highlighted that “over decades, the unstable alleles expand to more than ten times their initial length.” McCarroll noted that “these experiments have changed how we think about how Huntington’s develops.”
“It’s been known that these repeats expand in neurons,” said co-first author Seva Kashin of the Broad Institute. “But the ability to take a particular cell and measure both the CAG length and the transcriptional profile—that’s a really important underpinning that’s allowed for really powerful analysis.”
Preventing symptoms
“A lot was known about Huntington’s disease before we started this work, but there were gaps and inconsistencies in our collective understanding,” said Bob Handsaker, Broad Institute, a co-first author of the study. “We’ve been able to piece together the full trajectory of the pathology as it unfolds over decades in individual neurons, and that gives us potentially many different time points at which we can intervene therapeutically.”
The authors write that most current advanced therapies focus on reducing expression of HTT under the assumption that reducing the expression of the mutation will reduce symptoms and disease progression. This study offers an explanation for why those therapies have shown limited success. The authors concluded from their results that the disease pathology process occurs over the majority of the life of the affected neuron before cell death. This provides a longer time span for therapeutic applications.
The authors surmised, “If a cell spends 95% of its life in phase A, then even modestly slowing somatic expansion might substantially postpone HD symptom onset.” Treatments and therapies aimed at slowing the DNA-repeat expansion could postpone toxicity in many more cells, delaying or even preventing the onset of the disease. The authors also suggested that these therapies may be able to slow progression in individuals who already are showing symptoms, not just those who receive therapies early.
McCarroll said that the findings “will apply in DNA-repeat disorders beyond Huntington’s disease,” having broader implications for understanding related disorders caused by the expansion of repeat sequences, including fragile X syndrome and myotonic dystrophy.
While this study illuminated a mechanism for delayed onset of symptoms in patients having the mutation in HTT, many answers still lie in the dark. The inter-institution team is continuing to investigate remaining questions including why repeats expand more rapidly in some neurons than others and how the CAG repeats over 150 repeats trigger cell death. They hope that their findings will continue to shine light on possible interventions to slow the disease-driving process.
“It’s going to take much scientific work by many people to get to treatments that slow the expansion of DNA repeats,” McCarroll said. “But we’re hopeful that understanding this as the central disease-driving process leads to deep focus and new options.”
Finally, Berretta emphasized the importance of patient contributions to the research. “This would not have been possible without the altruism of many brain donors who have left a legacy of knowledge that will last and benefit many other people,” she said.