

In a pioneering study poised to reshape our understanding of addiction and brain aging, researchers at UTHealth Houston have unveiled compelling molecular evidence that substance use disorders (SUDs) accelerate biological aging within the human brain. Published in the April 29, 2025 issue of Genomic Psychiatry, this landmark research leverages brain-specific epigenetic clocks and advanced transcriptomic analyses to dissect the distinct yet convergent pathways through which substances such as alcohol, opioids, and stimulants hasten neural decline.
The investigation, spearheaded by Drs. Bruno Kluwe-Schiavon, Gabriel Fries, and Consuelo Walss-Bass, focused intently on the dorsolateral prefrontal cortex — a cerebral region integral to executive functions and decision-making, heavily implicated in the neuropathology of addiction. By examining postmortem brain tissues from 58 donors diagnosed with various SUDs, the team applied specialized epigenetic clocks tailored explicitly for cortical tissues. These include DNAmClockCortical, CerebralCortexClockcommon, and PCBrainAge, technologies that transcend previous methodologies by affording precision aging measurements rooted in neural epigenetic modifications.
This refined approach exposed nuanced molecular signatures that delineate how different classes of addictive substances uniquely disrupt neural aging. Alcohol use disorder (AUD) was associated with dysregulation in protein phosphorylation cascades, aberrant signal transduction pathways, and impairments in glutamatergic synaptic function — phenomena known to contribute to synaptic plasticity disruption and neurodegeneration. In contrast, opioid use disorder (OUD) exhibited alterations prominently in transcriptional regulation, neurodevelopmental gene networks, and immune-inflammatory signaling, highlighting the critical role of neuroimmune interactions in opioid-induced brain aging. Stimulant use disorder (StUD) unveiled a distinctive transcriptomic profile marked by oxidative stress responses, hypoxia-inducible factor (HIF) pathway activation, and modifications in cell adhesion mechanisms, reflecting a cellular environment under significant oxidative duress.
Despite these substance-specific pathways, the study illuminated several convergent mechanisms driving accelerated aging across the spectrum of SUDs. Notably, mitochondrial dysfunction emerged as a central theme, implicating compromised energy homeostasis and perturbed redox balance as key contributors to premature cellular senescence in neural tissues. Dr. Fries, co-corresponding author, emphasized how mitochondrial impairment synergizes with chronic neuroinflammation and oxidative stress to accelerate molecular decay, thereby shortening the biological lifespan of neurons independent of chronological age.
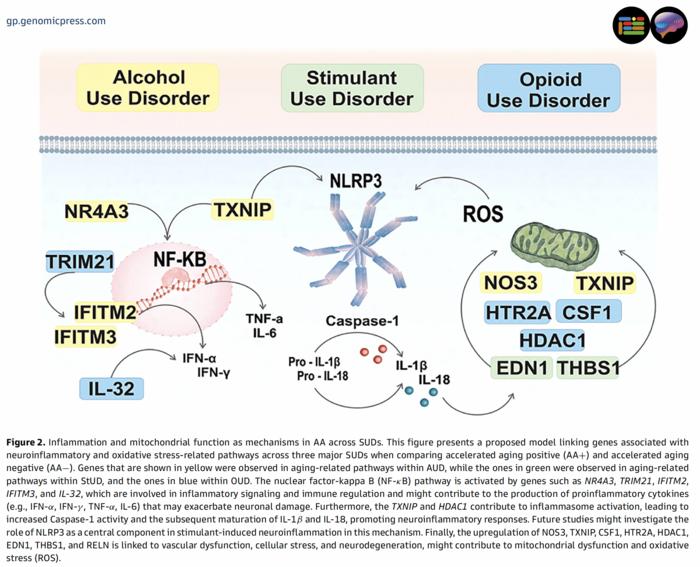
One particularly striking aspect of the analysis was the identification of neuroinflammatory cascades orchestrated through the nuclear factor-kappa B (NF-κB) signaling axis. Genes such as NR4A3, TRIM21, IFITM2, IFITM3, and IL-32 were found upregulated across SUD-affected brains, potentially driving enhanced production of proinflammatory cytokines including interferon-alpha, interferon-gamma, tumor necrosis factor-alpha, and interleukins 6, 1β, and 18. This persistent inflammatory milieu likely exacerbates synaptic dysfunction and neuronal loss, compounding the aging process at a cellular level.
Furthermore, inflammasome activation pathways — particularly involving TXNIP and HDAC1 — facilitate an increase in caspase-1 enzymatic activity, promoting the maturation and secretion of inflammatory interleukins that perpetuate neurodegenerative pathologies. Emerging evidence implicates NLRP3 as a potential linchpin in stimulant-induced neuroinflammation, a promising target for future mechanistic studies and therapeutic intervention aimed at attenuating accelerated aging in stimulant users.
Vascular dysfunction, a hallmark of neurodegeneration, was also underscored by the upregulation of multiple genes linked to endothelial integrity and cellular stress responses, including NOS3, CSF1, HTR2A, EDN1, THBS1, and RELN. These molecular disturbances may underlie microvascular compromise and oxidative stress, fostering an environment conducive to mitochondrial failure and exacerbated reactive oxygen species (ROS) production.
The clinical implications resonating from these findings are profound. The classical model of addiction as purely behavioral is challenged by data depicting SUDs as potent accelerants of neurobiological aging, implicating premature cortical exhaustion and cognitive decline as integral to relapse vulnerability. As Dr. Kluwe-Schiavon articulates, relapse may represent not just a lapse in willpower but a consequence of an aged and fatigued neural substrate.
This paradigm shift beckons the advent of a novel branch of psychiatry dedicated to understanding aging trajectories in young individuals afflicted by substance misuse. Longitudinal studies with integrated datasets encompassing methylation markers, transcriptomics, and neuroimaging biomarkers are essential to unravel the temporal dynamics of brain aging relative to exposure, remission, and relapse phases.
In his accompanying editorial, Dr. Julio Licinio eloquently frames the discourse, emphasizing that accelerated aging in SUDs is deeply rooted in molecular and epigenetic architecture rather than superficial or metaphorical alterations. He underscores the far-reaching consequences for public health strategies, criminal justice policies, and addiction treatment paradigms, urging a reevaluation of addiction through the lens of neurodegenerative disease acceleration rather than moral failing alone.
The authors acknowledge the study’s constraints, including a modest sample size and cross-sectional design, which currently limit the inference of causality. Nevertheless, this work lays an indispensable foundation for future large-scale investigations. Intriguingly, interindividual variability in aging speed under similar substance exposures raises pivotal questions regarding genetic susceptibility and the imprinting of early-life adversity as epigenetic scars influencing vulnerability.
Looking forward, therapeutic opportunities may emerge from interventions targeting mitochondrial preservation, anti-inflammatory modulation, and epigenetic rejuvenation. Dr. Licinio posits that anti-aging strategies, historically relegated to cosmetic and biohacking realms, might find their most urgent application in treating the neurobiological ravages of addiction, possibly heralding innovative avenues for recovery and prevention.
This groundbreaking study — “Deciphering the molecular basis of accelerated biological aging in substance use disorder: Integrative transcriptomic analysis” — is openly accessible in Genomic Psychiatry. It not only deepens our mechanistic understanding of addiction’s impact on the brain but also invites a holistic reframing of addiction as a disorder with profound implications on the biology of aging.
Subject of Research: People
Article Title: Deciphering the molecular basis of accelerated biological aging in substance use disorder: Integrative transcriptomic analysis
News Publication Date: 29-Apr-2025
Web References:
Research Article: https://doi.org/10.61373/gp025a.0029
Editorial Article: https://doi.org/10.61373/gp025d.0035
Image Credits: Consuelo Walss-Bass
Keywords: Senescence, Discovery research, Clinical research, Disordered regions, Regulation by phosphorylation, Genetic disorders, Neural pathways, HIF pathway, Molecular networks, Signaling networks, Adhesion signaling, Behavioral addiction, Genetic medicine, DNA regions, Genomic analysis
Tags: advanced methodologies in brain researchalcohol use disorder effectsbiological aging and substance usedorsolateral prefrontal cortex functionepigenetic clocks in neurosciencemolecular pathways of addictionneural decline and addictionneurobiology of decision-makingopioid impact on brain agingprecision measurements in health sciencessubstance use disorders and brain agingtranscriptomic analysis in addiction research

