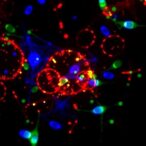
In a groundbreaking study emerging from The University of Texas MD Anderson Cancer Center, researchers have uncovered a novel molecular pathway that sheds light on the intricate relationship between vascular aging, disturbed blood flow, and the instability of atherosclerotic plaques—key contributors to heart attacks and strokes. This pioneering work advances our understanding of how cellular senescence in blood vessel walls disrupts vascular homeostasis and promotes dangerous thrombotic events.
The research focuses on endothelial cells, which form the inner lining of blood vessels and play a crucial role in maintaining vascular integrity. Under normal conditions, these cells regulate blood flow, prevent inflammation, and sustain a protective environment within arteries. However, this balance is disturbed when blood flow becomes irregular or turbulent, as seen in areas prone to plaque development. Such disturbed flow decreases levels of certain protective regulatory proteins, specifically LATS1 and LATS2, crucial components that help stabilize endothelial cell function.
What the investigators discovered is that the loss of LATS1/2 proteins triggers a shift in endothelial cells into a senescent state—a stressed and aging condition where cells cease to divide yet remain metabolically active. More alarmingly, this senescent state is not benign. The endothelial cells become hyperactive and dysfunctional, fostering a pro-inflammatory environment within the vascular wall. This senescent phenotype was shown to significantly upregulate the enzyme CD38, a critical factor that rewires cellular metabolism and energy utilization in these damaged cells.
CD38 activation within senescent endothelial cells emerges as a pivotal driver of plaque vulnerability. By altering the metabolic circuitry, CD38 enables these cells to ramp up their inflammatory responses, exacerbating vessel wall instability and promoting the formation of thrombi—blood clots that can occlude arteries and precipitate acute coronary syndromes or cerebrovascular accidents. Preclinical models demonstrated that overexpression of CD38 leads to marked inflammatory signaling, leaky blood vessels, and abnormal neovascularization—all hallmarks of high-risk plaques that can rupture suddenly.
Intriguingly, the team employed advanced molecular profiling and sophisticated in vitro and in vivo experiments to delineate this cascade. Genetic knockout models lacking LATS1/2 in endothelial cells recapitulated senescence-related phenotypes and elevated CD38 expression, corroborating the causal link. More compellingly, pharmacological inhibition of CD38 effectively reversed endothelial dysfunction and reduced thrombotic complications in these models, suggesting a promising therapeutic avenue.
The implications of this work extend beyond cardiovascular disease alone. The study offers a mechanistic insight into why certain cancer therapies, known to accelerate cellular aging and senescence, unexpectedly elevate cardiovascular risk. Many anticancer treatments induce senescence in both malignant and healthy tissues, potentially precipitating vascular inflammation and plaque destabilization via similar molecular pathways. Understanding this linkage opens the door to mitigating cardiovascular side effects in cancer patients, a population that faces unique vascular challenges.
Moreover, the findings underscore the intricate interplay between biomechanical forces such as shear stress and cellular metabolic reprogramming. Disturbed blood flow acts as a catalyst that signals endothelial cells to shift their metabolic state, mediated through the downregulation of LATS1/2 and subsequent CD38 upregulation. This cross-talk between physical forces and molecular signaling networks ultimately dictates vascular pathology progression.
From a therapeutic perspective, this research points toward repurposing existing FDA-approved CD38 inhibitors, currently used in oncology, to stabilize atherosclerotic plaques and prevent thrombotic cardiovascular events. Leveraging these drugs could provide a rapid translational pathway to clinical interventions aimed at reducing heart attacks and strokes by targeting the senescence-metabolism axis unveiled in this study.
Importantly, the investigators validated their findings using human carotid plaque samples, confirming that the molecular signatures identified in animal models are present in human disease. This translational relevance enhances the potential impact of the discovery and sets the stage for future clinical trials.
While the study provides compelling evidence linking endothelial senescence, CD38 activation, and thrombotic risk, the authors stress that further research is needed to identify reliable biomarkers that could predict individuals at higher risk of plaque rupture or cardiovascular events. Such biomarkers could enable earlier intervention and personalized treatment strategies.
In conclusion, this work from MD Anderson Cancer Center elegantly elucidates how cellular aging within blood vessels, driven by mechanical disturbances and metabolic alterations, contributes to cardiovascular disease pathogenesis. The identification of CD38 as a molecular lynchpin in this process reveals novel opportunities for targeted therapy, offering hope for millions affected by vascular disease and cancer treatment-related cardiovascular complications.
—
Subject of Research: Molecular mechanisms linking endothelial senescence, CD38 activation, and atherosclerotic plaque instability
Article Title: Newly Discovered Molecular Pathway Links Endothelial Cell Senescence to Plaque Instability and Thrombosis
News Publication Date: June 4, 2026
Web References:
– University of Texas MD Anderson Cancer Center: https://www.mdanderson.org/
– Circulation Research Article: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.125.327427
Image Credits: The University of Texas MD Anderson Cancer Center
Keywords: Endothelial cells, cellular senescence, atherosclerosis, CD38 enzyme, LATS1/2 proteins, vascular inflammation, disturbed blood flow, thrombosis, plaque instability, cardiovascular disease, cancer treatment side effects, metabolic reprogramming
Tags: blood vessel homeostasis disruptioncellular mechanisms of thrombosisdisturbed blood flow and plaque instabilityendothelial cell senescence and heart diseaseendothelial dysfunction and cardiovascular riskendothelial senescence and thrombotic eventsimpact of senescent cells on blood vesselsinflammation in atherosclerotic plaque developmentLATS1 and LATS2 protein functionmolecular pathways in vascular healthrole of endothelial cells in strokevascular aging and atherosclerosis