

In a groundbreaking study recently published in the prestigious journal Proceedings of the National Academy of Sciences (PNAS), an international team of scientists from Mainz, Valencia, Madrid, and Zurich have unveiled a transformative perspective on one of biology’s greatest enigmas: the emergence of the eukaryotic cell. This event, marking the most profound increase in cellular complexity in Earth’s evolutionary timeline, has long been shrouded in mystery, largely due to the absence of evolutionary intermediates bridging the gap between the simpler prokaryotic organisms and the sophisticated eukaryotes. This gap, often referred to as the ‘black hole at the heart of biology,’ has challenged researchers for decades. Through a careful integration of computational modeling, evolutionary theory, and quantitative analyses, these researchers offer an elegant and compelling model describing how genetic architectures evolved to support this leap in complexity.
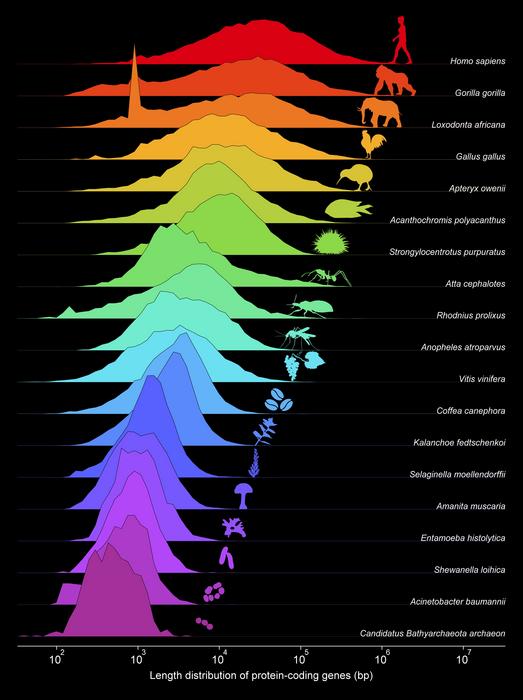
The team’s approach hinges upon a comprehensive analysis of gene and protein length distributions over evolutionary time. Using an extensive dataset comprising nearly 10,000 proteomes and over 33,000 genomes spanning all domains of life, the researchers demonstrate that gene and protein lengths consistently follow log-normal distributions—a hallmark of multiplicative stochastic processes. Log-normal distributions, common throughout natural and social sciences, suggest that gene length evolution does not occur via simple linear increments but through multiplicative growth mechanisms, likely influenced by various genetic operators acting in unison. By modeling gene length evolution as a multiplicative stochastic phenomenon, the researchers provide a quantitative framework that captures the underlying dynamics shaping the genetic complexity of life.
At the core of their findings is the observation that average gene lengths have evolved in an exponential trajectory starting from the Last Universal Common Ancestor (LUCA), the hypothesized root from which all current life forms—the Bacteria, Archaea, and Eukarya—descended. Notably, the researchers identified a scaling-invariant mechanism governing gene growth, whereby the variance in gene length distributions correlates directly to the mean protein length regardless of species lineage. This discovery implies a universal evolutionary dynamic, transcending vast phylogenetic distances, and offers a robust metric for assessing organismal complexity. Indeed, from a single statistic—the average length of protein-coding genes—one can infer the full distribution of gene lengths within a species, underscoring the power of this framework.
However, this growth in gene length and corresponding protein length does not continue indefinitely in tandem. In prokaryotes, genes are predominantly coding sequences with minimal non-coding regions, causing gene and protein lengths to evolve synchronously. Yet, as the average gene length approaches approximately 1,500 nucleotides, this relationship diverges sharply. Beyond this threshold, the average protein length plateaus at about 500 amino acids, signaling the rise of the eukaryotic cell. From this point onward, gene length continues to increase markedly due to the accumulation and expansion of non-coding sequences — introns and regulatory elements within genes — which play a pivotal role in eukaryotic gene regulation and complexity. This bifurcation marks a critical juncture in evolutionary history where genomic content and architecture undergo profound shifts.
The researchers further probe this critical point via a rigorous analysis rooted in physics, particularly the study of phase transitions analogous to those observed in magnetic materials. They reveal that the evolution of gene length crosses a threshold at 1,500 nucleotides, reminiscent of a critical phase transition that demarcates two distinct evolutionary regimes: a ‘coding phase,’ dominated by prokaryotic lineages, and a ‘non-coding phase,’ characteristic of eukaryotic organisms. This phase transition is not merely metaphorical but manifests algorithmically within the genetic architecture, influencing the fundamental computational operations of gene expression and protein synthesis.
An intriguing aspect of this transition is the phenomenon known as critical slowing down. Borrowed from statistical physics, critical slowing down describes the system’s propensity to become trapped in numerous metastable states near the critical point—akin to evolutionary stasis or slow adaptation. Observations in early protists and fungi confirm this dynamic, reflecting complex evolutionary constraints operating during eukaryogenesis. Such a slowing could explain periods of apparent evolutionary “bottleneck,” emphasizing the challenges intrinsic to transitioning the molecular machinery from prokaryotic simplicity towards eukaryotic sophistication.
Professor Jordi Bascompte from the University of Zurich elucidates the algorithmic essence of this transition. During the coding phase, resembling LUCA-like conditions with relatively short proteins, increasing protein length and corresponding gene length was computationally straightforward—akin to a linear search problem. However, as proteins extended, the combinatorial complexity associated with finding viable, longer proteins increased exponentially, rendering the ‘search’ biologically and computationally untenable. This impasse was resolved abruptly and innovatively at the phase transition point through the incorporation of extensive non-coding sequences. These non-coding regions facilitated the emergence of the spliceosome and the segregation of genetic processes across a nucleus, effectively partitioning transcription and splicing from translation. Such architectural innovations significantly lowered the computational complexity of protein synthesis, enabling the curated expression of functional proteins from longer, more complex genes.
This algorithmic phase transition, dated to approximately 2.6 billion years ago, signified a revolutionary milestone in the evolution of life. The eukaryotic cell emerged not by mere incremental modifications but through a fundamental transformation in the organizational and computational principles underlying genetic information processing. This paradigm shift unlocked a cascade of subsequent evolutionary milestones, including multicellularity, sexual reproduction, and complex social behaviors—cornerstones that have shaped terrestrial life into its present diversity.
The interdisciplinary nature of this study marks a critical progression in evolutionary biology. By synergizing concepts from computational biology, quantitative evolutionary theory, and statistical physics, the authors transcend traditional disciplinary boundaries. Their work invites further exploration into related domains, such as information theory and energy dynamics in biological systems, flagging new frontiers in understanding life’s complexity from a theoretical vantage point. Dr. Enrique M. Muro of Johannes Gutenberg University Mainz, one of the project’s representatives, emphasizes the broad appeal and potential impact of this research, predicting it will stimulate a multiplicity of interdisciplinary investigations into the evolutionary origins and algorithms embedded within living systems.
This study does more than just decode the past; it reframes our conceptualization of life’s grand transitions as complex, algorithmically defined events rather than gradual, stochastic processes. Through this lens, life’s history becomes a narrative of computational optimization and critical threshold crossings, entwined with physical laws governing phase transitions. Importantly, the research underscores the power of quantitative biology to unravel intricate evolutionary enigmas by harnessing mathematical rigor and computational insights, thereby redefining our understanding of biological innovation.
In sum, the emergence of the eukaryotic cell represents an evolutionary algorithmic phase transition—a bifurcation demarked both by genomic composition and by the underlying computational architectures that enable life’s increasing complexity. This discovery not only fills an essential gap in evolutionary theory but also establishes a conceptual framework for future studies aimed to decode the deep history of life’s major transitions. As such, this research heralds a new era in evolutionary biology where computational principles illuminate the enigmatic origins of complex life forms on Earth.
Subject of Research: The evolutionary origin and complexity increase of the eukaryotic cell through gene and protein length distribution analysis modeled as an evolutionary algorithmic phase transition.
Article Title: The emergence of eukaryotes as an evolutionary algorithmic phase transition
News Publication Date: 27-Mar-2025
Web References: http://dx.doi.org/10.1073/pnas.2422968122
Image Credits: ill./©: Fernando J. Ballesteros
Keywords: eukaryotic cell, evolutionary biology, gene length distribution, protein length, phase transition, algorithmic evolution, LUCA, multiplicative stochastic processes, genome complexity, computational biology
Tags: black hole of biologybridging prokaryotic and eukaryotic lifecomputational modeling in evolutionemergence of complex cellseukaryotic cell evolutionevolutionary algorithms in biologygene and protein length distributionsgenetic architectures and complexityinterdisciplinary approaches in scienceproteome dataset analysisquantitative analysis in evolutionary studiestransformative biological transitions

